|
|
PDBPARSE documentation |
/homes/user/test/data/structure/1cs4.ent SEQRESLENDIF 1 (A) ATOMCOL12 429 BADINDEX 1 (A) GAPPEDOK 1 (A) SECSTART 1 1 ILE 384 SECSTART 1 1 ILE 384 // /homes/user/test/data/structure/1ii7.ent SEQRESLENDIF 1 (A) ATOMCOL12 390 SECBOTH 1 1 SER 57 GLU 73 SECBOTH 1 1 VAL 78 ILE 81 SECBOTH 1 1 LYS 2 LEU 6 // /homes/user/test/data/structure/2hhb.ent ATOMCOL12 1277 // |
ID 1cs4
XX
DE MOL_ID: 1; MOLECULE: TYPE V ADENYLATE CYCLASE;
XX
OS MOL_ID: 1; ORGANISM_SCIENTIFIC: CANIS FAMILIARIS;
XX
EX METHOD xray; RESO 2.50; NMOD 1; NCHN 1; NGRP 0;
XX
CN [1]
XX
IN ID A; NR 52; NL 7; NH 0; NE 0;
XX
SQ SEQUENCE 52 AA; 5817 MW; D8CCAE0E1FC0849A CRC64;
ADIEGFTSLA SQCTAQELVM TLNELFARFD KLAAENHCLR IKILGDCYYC VS
XX
RE 1 1 2 396 D ASP . . . . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 3 397 I ILE . . . . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 4 398 E GLU . . . . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 5 399 G GLY 1 1 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 6 400 F PHE 1 1 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 7 401 T THR 1 1 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 8 402 S SER 1 1 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 9 403 L LEU 1 1 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 10 404 A ALA 1 1 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 11 405 S SER 1 1 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 12 406 Q GLN . . . . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 13 407 C CYS . . . . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 14 408 T THR 2 2 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 15 409 A ALA 2 2 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 16 410 Q GLN 2 2 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 17 411 E GLU 2 2 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 18 412 L LEU 2 2 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 19 413 V VAL 2 2 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 20 414 M MET 2 2 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 21 415 T THR 2 2 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 22 416 L LEU 2 2 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 23 417 N ASN 2 2 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 24 418 E GLU 2 2 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 25 419 L LEU 2 2 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 26 420 F PHE 2 2 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 27 421 A ALA 2 2 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 28 422 R ARG 2 2 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 29 423 F PHE 2 2 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 30 424 D ASP 2 2 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 31 425 K LYS 2 2 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 32 426 L LEU 2 2 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 33 427 A ALA 2 2 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 34 428 A ALA 2 2 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 35 429 E GLU 2 2 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 36 430 N ASN 2 2 H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
[Part of this file has been deleted for brevity]
AT 1 1 5 . 1002 . FOK H C9 42.200 -11.309 50.489 1.00 41.39
AT 1 1 5 . 1002 . FOK H O6 42.275 -12.455 49.593 1.00 43.23
AT 1 1 5 . 1002 . FOK H C10 43.008 -11.601 51.811 1.00 39.11
AT 1 1 5 . 1002 . FOK H C11 40.680 -11.078 50.616 1.00 44.36
AT 1 1 5 . 1002 . FOK H O7 40.106 -10.945 51.688 1.00 48.77
AT 1 1 5 . 1002 . FOK H C12 39.943 -11.046 49.301 1.00 40.67
AT 1 1 5 . 1002 . FOK H C13 40.595 -10.085 48.292 1.00 41.47
AT 1 1 5 . 1002 . FOK H C14 40.276 -10.620 46.930 1.00 46.69
AT 1 1 5 . 1002 . FOK H C15 39.971 -11.751 46.590 1.00 53.22
AT 1 1 5 . 1002 . FOK H C16 40.047 -8.685 48.426 1.00 42.42
AT 1 1 5 . 1002 . FOK H C17 42.671 -8.737 50.253 1.00 39.67
AT 1 1 5 . 1002 . FOK H C18 46.732 -13.026 51.827 1.00 35.74
AT 1 1 5 . 1002 . FOK H C19 45.859 -11.483 53.586 1.00 34.48
AT 1 1 5 . 1002 . FOK H C20 42.913 -10.426 52.807 1.00 39.44
AT 1 1 5 . 1002 . FOK H C21 45.883 -9.553 47.821 1.00 42.15
AT 1 1 5 . 1002 . FOK H O5 46.157 -10.520 47.166 1.00 40.91
AT 1 1 5 . 1002 . FOK H C22 46.769 -8.315 48.006 1.00 37.08
AT 1 1 6 . 1003 . MES H O1 45.676 7.326 49.092 1.00 77.86
AT 1 1 6 . 1003 . MES H C2 44.367 6.816 48.900 1.00 75.17
AT 1 1 6 . 1003 . MES H C3 44.349 5.317 48.923 1.00 74.42
AT 1 1 6 . 1003 . MES H N4 44.832 4.804 50.196 1.00 72.45
AT 1 1 6 . 1003 . MES H C5 46.234 5.425 50.473 1.00 73.23
AT 1 1 6 . 1003 . MES H C6 46.176 6.914 50.355 1.00 75.06
AT 1 1 6 . 1003 . MES H C7 44.806 3.336 50.302 1.00 73.39
AT 1 1 6 . 1003 . MES H C8 44.672 2.791 51.713 1.00 76.85
AT 1 1 6 . 1003 . MES H S 45.724 1.379 51.967 1.00 78.26
AT 1 1 6 . 1003 . MES H O1S 47.062 1.828 51.737 1.00 79.39
AT 1 1 6 . 1003 . MES H O2S 45.303 0.380 51.016 1.00 81.58
AT 1 1 6 . 1003 . MES H O3S 45.523 0.961 53.326 1.00 80.59
AT 1 1 6 . 1004 . MES H O1 59.246 -5.152 27.381 1.00 99.99
AT 1 1 6 . 1004 . MES H C2 60.067 -4.021 27.127 1.00 99.99
AT 1 1 6 . 1004 . MES H C3 60.447 -3.301 28.378 1.00 99.78
AT 1 1 6 . 1004 . MES H N4 61.180 -4.156 29.270 1.00 96.33
AT 1 1 6 . 1004 . MES H C5 60.358 -5.461 29.506 1.00 97.90
AT 1 1 6 . 1004 . MES H C6 59.965 -6.072 28.203 1.00 99.68
AT 1 1 6 . 1004 . MES H C7 61.596 -3.484 30.507 1.00 93.33
AT 1 1 6 . 1004 . MES H C8 61.931 -2.010 30.442 1.00 90.74
AT 1 1 6 . 1004 . MES H S 60.763 -0.978 31.301 0.50 90.72
AT 1 1 6 . 1004 . MES H O1S 59.476 -1.170 30.680 0.50 91.60
AT 1 1 6 . 1004 . MES H O2S 61.249 0.383 31.164 0.50 91.20
AT 1 1 6 . 1004 . MES H O3S 60.776 -1.430 32.647 0.50 90.05
AT 1 1 7 . 1005 . POP H P1 58.812 -7.766 57.091 1.00 57.40
AT 1 1 7 . 1005 . POP H O1 60.254 -7.589 56.745 1.00 54.93
AT 1 1 7 . 1005 . POP H O2 58.618 -8.839 58.095 1.00 55.36
AT 1 1 7 . 1005 . POP H O3 57.949 -8.024 55.908 1.00 55.10
AT 1 1 7 . 1005 . POP H O 58.295 -6.370 57.759 1.00 57.30
AT 1 1 7 . 1005 . POP H P2 56.998 -5.955 58.661 1.00 59.66
AT 1 1 7 . 1005 . POP H O4 57.491 -5.746 60.070 1.00 54.95
AT 1 1 7 . 1005 . POP H O5 56.004 -7.075 58.550 1.00 56.24
AT 1 1 7 . 1005 . POP H O6 56.427 -4.710 58.044 1.00 56.50
//
|
ID 1ii7
XX
DE MOL_ID: 1; MOLECULE: MRE11 NUCLEASE;
XX
OS MOL_ID: 1; ORGANISM_SCIENTIFIC: PYROCOCCUS FURIOSUS;
XX
EX METHOD xray; RESO 2.20; NMOD 1; NCHN 1; NGRP 0;
XX
CN [1]
XX
IN ID A; NR 65; NL 6; NH 0; NE 0;
XX
SQ SEQUENCE 65 AA; 7395 MW; 75FBE75B22FD3678 CRC64;
MKFAHLADIH LGYEQFHKPQ REEEFAEAFK NALEIAVQEN VDFILIAGDL FHSSRPSPGT
LKKAI
XX
RE 1 1 8 8 D ASP . . . . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 9 9 I ILE . . . . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 10 10 H HIS . . . . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 11 11 L LEU . . . . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 12 12 G GLY . . . . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 13 13 Y TYR . . . . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 14 14 E GLU 1 1 H 5 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 15 15 Q GLN 1 1 H 5 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 16 16 F PHE 1 1 H 5 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 17 17 H HIS 1 1 H 5 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 18 18 K LYS 2 A E . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 19 19 P PRO 2 A E . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 20 20 Q GLN 2 A E . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 21 21 R ARG 2 A E . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 22 22 E GLU 2 A E . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 23 23 E GLU 2 A E . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 24 24 E GLU 2 A E . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 25 25 F PHE 2 A E . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 26 26 A ALA 2 A E . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 27 27 E GLU 2 A E . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 28 28 A ALA 2 A E . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 29 29 F PHE 2 A E . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 30 30 K LYS 2 A E . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 31 31 N ASN 2 A E . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 32 32 A ALA 2 A E . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 33 33 L LEU 2 A E . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 34 34 E GLU 2 A E . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 35 35 I ILE 2 A E . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 36 36 A ALA 2 A E . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 37 37 V VAL 2 A E . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 38 38 Q GLN 2 A E . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 39 39 E GLU 2 A E . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 40 40 N ASN . . . . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 41 41 V VAL . . . . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
[Part of this file has been deleted for brevity]
AT 1 1 . 50 50 L LEU P CD2 12.425 39.035 22.798 1.00 23.77
AT 1 1 1 . 402 . PO4 H P 34.178 32.996 46.387 1.00 60.84
AT 1 1 1 . 402 . PO4 H O1 35.146 33.243 45.291 1.00 57.95
AT 1 1 1 . 402 . PO4 H O2 34.912 32.751 47.670 1.00 59.15
AT 1 1 1 . 402 . PO4 H O3 33.291 34.184 46.538 1.00 58.92
AT 1 1 1 . 402 . PO4 H O4 33.352 31.796 46.060 1.00 61.86
AT 1 1 2 . 403 . MN H MN 8.130 27.788 21.899 1.00 36.09
AT 1 1 2 . 404 . MN H MN 5.801 27.935 24.271 1.00 39.57
AT 1 1 3 . 405 . MN H MN 36.023 34.916 44.253 1.00 39.52
AT 1 1 3 . 406 . MN H MN 33.658 36.365 46.296 1.00 33.69
AT 1 1 5 . 501 . SO4 H S 17.175 28.112 32.476 1.00 100.80
AT 1 1 5 . 501 . SO4 H O1 18.136 28.230 31.357 1.00 100.18
AT 1 1 5 . 501 . SO4 H O2 17.097 26.692 32.887 1.00 100.80
AT 1 1 5 . 501 . SO4 H O3 17.633 28.926 33.626 1.00 100.14
AT 1 1 5 . 501 . SO4 H O4 15.834 28.575 32.045 1.00 100.56
AT 1 1 5 . 502 . SO4 H S 0.566 29.512 36.007 1.00 86.73
AT 1 1 5 . 502 . SO4 H O1 1.690 28.556 35.971 1.00 87.27
AT 1 1 5 . 502 . SO4 H O2 -0.620 28.803 36.523 1.00 87.87
AT 1 1 5 . 502 . SO4 H O3 0.896 30.642 36.905 1.00 86.58
AT 1 1 5 . 502 . SO4 H O4 0.287 30.037 34.658 1.00 86.51
AT 1 1 5 . 503 . SO4 H S -13.586 39.644 36.031 1.00 100.28
AT 1 1 5 . 503 . SO4 H O1 -12.340 39.512 35.250 1.00 100.72
AT 1 1 5 . 503 . SO4 H O2 -14.638 38.811 35.421 1.00 100.46
AT 1 1 5 . 503 . SO4 H O3 -13.347 39.201 37.420 1.00 99.66
AT 1 1 5 . 503 . SO4 H O4 -14.020 41.056 36.015 1.00 99.97
AT 1 1 6 . 401 . 101 H P 7.599 25.305 23.994 1.00 56.33
AT 1 1 6 . 401 . 101 H O1P 8.249 24.467 25.030 1.00 56.70
AT 1 1 6 . 401 . 101 H O2P 6.700 26.285 24.649 1.00 54.49
AT 1 1 6 . 401 . 101 H O3P 8.637 26.026 23.216 1.00 53.97
AT 1 1 6 . 401 . 101 H O5* 7.095 23.970 23.128 1.00 59.20
AT 1 1 6 . 401 . 101 H C5* 7.073 23.961 21.762 1.00 66.74
AT 1 1 6 . 401 . 101 H C4* 6.041 23.013 21.296 1.00 71.22
AT 1 1 6 . 401 . 101 H O4* 6.029 21.855 22.189 1.00 73.78
AT 1 1 6 . 401 . 101 H C3* 4.736 23.676 21.350 1.00 73.80
AT 1 1 6 . 401 . 101 H O3* 4.355 23.874 19.995 1.00 76.51
AT 1 1 6 . 401 . 101 H C2* 3.864 22.749 22.165 1.00 74.04
AT 1 1 6 . 401 . 101 H C1* 4.682 21.474 22.506 1.00 74.70
AT 1 1 6 . 401 . 101 H N9 4.578 21.123 23.969 1.00 76.71
AT 1 1 6 . 401 . 101 H C8 3.630 21.533 24.876 1.00 76.87
AT 1 1 6 . 401 . 101 H N7 3.758 21.069 26.081 1.00 77.50
AT 1 1 6 . 401 . 101 H C5 4.896 20.300 25.989 1.00 77.78
AT 1 1 6 . 401 . 101 H C6 5.570 19.479 26.941 1.00 78.16
AT 1 1 6 . 401 . 101 H N6 5.155 19.409 28.200 1.00 78.77
AT 1 1 6 . 401 . 101 H N1 6.682 18.805 26.554 1.00 78.32
AT 1 1 6 . 401 . 101 H C2 7.090 18.888 25.277 1.00 78.14
AT 1 1 6 . 401 . 101 H N3 6.541 19.611 24.271 1.00 78.05
AT 1 1 6 . 401 . 101 H C4 5.403 20.288 24.700 1.00 78.10
AT 1 . . . 407 . HOH W O 5.997 27.242 22.189 1.00 38.84
AT 1 . . . 408 . HOH W O 35.697 35.756 46.350 1.00 41.39
AT 1 . . . 600 . HOH W O 20.825 31.690 27.031 1.00 20.90
//
|
ID 2hhb
XX
DE HEMOGLOBIN (DEOXY)
XX
OS HUMAN (HOMO SAPIENS)
XX
EX METHOD xray; RESO 1.74; NMOD 1; NCHN 4; NGRP 0;
XX
CN [1]
XX
IN ID A; NR 141; NL 1; NH 0; NE 0;
XX
SQ SEQUENCE 141 AA; 15126 MW; 34D13618E62A33C1 CRC64;
VLSPADKTNV KAAWGKVGAH AGEYGAEALE RMFLSFPTTK TYFPHFDLSH GSAQVKGHGK
KVADALTNAV AHVDDMPNAL SALSDLHAHK LRVDPVNFKL LSHCLLVTLA AHLPAEFTPA
VHASLDKFLA SVSTVLTSKY R
XX
CN [2]
XX
IN ID B; NR 146; NL 1; NH 0; NE 0;
XX
SQ SEQUENCE 146 AA; 15867 MW; EACBC707CFD466A1 CRC64;
VHLTPEEKSA VTALWGKVNV DEVGGEALGR LLVVYPWTQR FFESFGDLST PDAVMGNPKV
KAHGKKVLGA FSDGLAHLDN LKGTFATLSE LHCDKLHVDP ENFRLLGNVL VCVLAHHFGK
EFTPPVQAAY QKVVAGVANA LAHKYH
XX
CN [3]
XX
IN ID C; NR 141; NL 1; NH 0; NE 0;
XX
SQ SEQUENCE 141 AA; 15126 MW; 34D13618E62A33C1 CRC64;
VLSPADKTNV KAAWGKVGAH AGEYGAEALE RMFLSFPTTK TYFPHFDLSH GSAQVKGHGK
KVADALTNAV AHVDDMPNAL SALSDLHAHK LRVDPVNFKL LSHCLLVTLA AHLPAEFTPA
VHASLDKFLA SVSTVLTSKY R
XX
CN [4]
XX
IN ID D; NR 146; NL 2; NH 0; NE 0;
XX
SQ SEQUENCE 146 AA; 15867 MW; EACBC707CFD466A1 CRC64;
VHLTPEEKSA VTALWGKVNV DEVGGEALGR LLVVYPWTQR FFESFGDLST PDAVMGNPKV
KAHGKKVLGA FSDGLAHLDN LKGTFATLSE LHCDKLHVDP ENFRLLGNVL VCVLAHHFGK
EFTPPVQAAY QKVVAGVANA LAHKYH
XX
RE 1 1 1 1 V VAL . . . . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 2 2 L LEU . . . . . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 3 3 S SER 1 AA H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 4 4 P PRO 1 AA H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 5 5 A ALA 1 AA H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
RE 1 1 6 6 D ASP 1 AA H 1 . . 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00 0.00
[Part of this file has been deleted for brevity]
AT 1 . . . 174 . HOH W O -4.764 -6.228 5.515 8.00 40.89
AT 1 . . . 175 . HOH W O 23.809 19.925 1.758 8.00 39.37
AT 1 . . . 176 . HOH W O -7.871 -9.078 2.406 8.00 43.37
AT 1 . . . 177 . HOH W O 4.693 12.083 7.558 8.00 40.24
AT 1 . . . 178 . HOH W O 8.775 -23.438 16.055 8.00 42.33
AT 1 . . . 179 . HOH W O -7.480 -10.898 17.998 8.00 38.06
AT 1 . . . 180 . HOH W O -4.731 16.453 2.295 8.00 36.37
AT 1 . . . 181 . HOH W O -1.055 11.866 -0.448 8.00 43.19
AT 1 . . . 182 . HOH W O -27.610 -10.991 5.353 8.00 43.46
AT 1 . . . 183 . HOH W O 26.015 11.766 5.159 8.00 40.95
AT 1 . . . 184 . HOH W O -18.517 -8.355 15.267 8.00 35.55
AT 1 . . . 185 . HOH W O -14.034 2.806 -30.367 8.00 41.77
AT 1 . . . 186 . HOH W O -32.905 -9.033 0.480 8.00 43.68
AT 1 . . . 187 . HOH W O -28.749 -13.315 1.938 8.00 45.36
AT 1 . . . 188 . HOH W O 0.516 -8.074 -26.354 8.00 41.53
AT 1 . . . 189 . HOH W O -20.080 -9.873 -22.862 8.00 36.25
AT 1 . . . 190 . HOH W O -13.442 9.778 -13.572 8.00 39.70
AT 1 . . . 191 . HOH W O -24.804 -2.608 -15.488 8.00 37.79
AT 1 . . . 192 . HOH W O 6.547 9.706 16.296 8.00 41.86
AT 1 . . . 193 . HOH W O 0.029 22.606 14.164 8.00 43.02
AT 1 . . . 194 . HOH W O -11.367 0.306 28.463 8.00 44.30
AT 1 . . . 195 . HOH W O -19.950 -10.635 14.301 8.00 40.17
AT 1 . . . 196 . HOH W O -7.047 -6.324 20.098 8.00 36.98
AT 1 . . . 197 . HOH W O -23.876 1.108 14.102 8.00 33.31
AT 1 . . . 198 . HOH W O -34.199 8.033 11.037 8.00 40.72
AT 1 . . . 199 . HOH W O -14.173 13.393 -8.778 8.00 43.21
AT 1 . . . 200 . HOH W O 11.388 -11.044 24.763 8.00 39.34
AT 1 . . . 201 . HOH W O 3.735 -3.643 2.734 8.00 42.17
AT 1 . . . 202 . HOH W O 3.149 -0.692 2.083 8.00 41.40
AT 1 . . . 203 . HOH W O 4.511 -25.886 13.006 8.00 39.83
AT 1 . . . 204 . HOH W O 8.712 -21.655 3.577 8.00 43.08
AT 1 . . . 205 . HOH W O 22.926 -4.304 24.079 8.00 38.10
AT 1 . . . 206 . HOH W O 11.435 9.654 20.618 8.00 40.23
AT 1 . . . 207 . HOH W O 18.099 5.542 27.744 8.00 39.03
AT 1 . . . 208 . HOH W O 12.174 9.951 9.804 8.00 44.34
AT 1 . . . 209 . HOH W O 24.745 -2.501 15.270 8.00 39.78
AT 1 . . . 210 . HOH W O 24.231 0.100 14.764 8.00 42.94
AT 1 . . . 211 . HOH W O 23.324 -18.136 10.981 8.00 53.60
AT 1 . . . 212 . HOH W O 25.576 -22.211 6.309 8.00 45.18
AT 1 . . . 213 . HOH W O 14.639 24.823 -4.300 8.00 41.35
AT 1 . . . 214 . HOH W O 14.903 5.393 -23.047 8.00 37.45
AT 1 . . . 215 . HOH W O 16.650 -5.137 -16.717 8.00 39.12
AT 1 . . . 216 . HOH W O 7.424 -6.700 -20.085 8.00 38.62
AT 1 . . . 217 . HOH W O -1.263 -2.837 -21.251 8.00 45.10
AT 1 . . . 218 . HOH W O 23.120 -3.118 -12.992 8.00 37.05
AT 1 . . . 219 . HOH W O 23.664 0.968 -14.389 8.00 36.25
AT 1 . . . 220 . HOH W O 25.698 7.981 -15.362 8.00 35.85
AT 1 . . . 221 . HOH W O 30.009 16.347 -6.794 8.00 37.62
AT 1 . . . 222 . HOH W O 27.728 16.677 -1.376 8.00 42.54
AT 1 . . . 223 . HOH W O 8.142 18.836 1.041 8.00 39.90
//
|
Parses PDB files and writes protein CCF files.
Version: EMBOSS:6.3.0
Standard (Mandatory) qualifiers:
[-pdbpath] dirlist [./] This option specifies the location of
PDB files (input). A PDB file contains
protein coordinate and other data. A
detailed explanation of the PDB file format
is available on the PDB web site
http://www.rcsb.org/pdb/info.html.
-camask boolean [N] This option specifies whether to to mask
non-amino acid groups in protein chains
that do not contain a C-alpha atom. If
masked, the group will not appear in either
the CO or SQ records of the clean coordinate
file.
-camaska boolean [N] This option specifies whether to mask
amino acids in protein chains that do not
contain a C-alpha atom. If masked, the amino
acid will not appear in the CO record but
will still be present in the SQ record of
the clean coordinate file.
-atommask boolean [N] This option specifies whether to mask
amino acid residues in protein chains with a
single atom only. If masked, the amino acid
will appear not appear in the CO record but
will still be present in the SQ record of
the clean coordinate file.
[-ccfoutdir] outdir [./] This option specifies the location of
CCF files (clean coordinate files) (output).
A 'protein clean cordinate file' contains
protein coordinate and other data for a
single PDB file. The files, generated by
using PDBPARSE, are in CCF format
(EMBL-like) and contain 'cleaned-up' data
that is self-consistent and error-corrected.
Records for residue solvent accessibility
and secondary structure are added to the
file by using PDBPLUS.
-logfile outfile [pdbparse.log] This option specifies tame of
the log file for the build. The log file
may contain messages about inconsistencies
or errors in the PDB files that were parsed.
Additional (Optional) qualifiers:
-[no]ccfnaming boolean [Y] This option specifies whether to use
pdbid code to name the output files. If set,
the PDB identifier code (from the PDB file)
is used to name the file. Otherwise, the
output files have the same names as the
input files.
-chnsiz integer [5] Minimum number of amino acid residues in
a chain for it to be parsed. (Any integer
value)
-maxmis integer [3] Maximum number of permissible mismatches
between the ATOM and SEQRES sequences. (Any
integer value)
-maxtrim integer [10] Max. no. residues to trim when checking
for missing C-terminal SEQRES sequences.
(Any integer value)
Advanced (Unprompted) qualifiers: (none)
Associated qualifiers:
"-pdbpath" associated qualifiers
-extension1 string Default file extension
"-ccfoutdir" associated qualifiers
-extension2 string Default file extension
"-logfile" associated qualifiers
-odirectory string Output directory
General qualifiers:
-auto boolean Turn off prompts
-stdout boolean Write first file to standard output
-filter boolean Read first file from standard input, write
first file to standard output
-options boolean Prompt for standard and additional values
-debug boolean Write debug output to program.dbg
-verbose boolean Report some/full command line options
-help boolean Report command line options and exit. More
information on associated and general
qualifiers can be found with -help -verbose
-warning boolean Report warnings
-error boolean Report errors
-fatal boolean Report fatal errors
-die boolean Report dying program messages
-version boolean Report version number and exit
|
| Qualifier | Type | Description | Allowed values | Default |
|---|---|---|---|---|
| Standard (Mandatory) qualifiers | ||||
| [-pdbpath] (Parameter 1) |
dirlist | This option specifies the location of PDB files (input). A PDB file contains protein coordinate and other data. A detailed explanation of the PDB file format is available on the PDB web site http://www.rcsb.org/pdb/info.html. | Directory with files | ./ |
| -camask | boolean | This option specifies whether to to mask non-amino acid groups in protein chains that do not contain a C-alpha atom. If masked, the group will not appear in either the CO or SQ records of the clean coordinate file. | Boolean value Yes/No | No |
| -camaska | boolean | This option specifies whether to mask amino acids in protein chains that do not contain a C-alpha atom. If masked, the amino acid will not appear in the CO record but will still be present in the SQ record of the clean coordinate file. | Boolean value Yes/No | No |
| -atommask | boolean | This option specifies whether to mask amino acid residues in protein chains with a single atom only. If masked, the amino acid will appear not appear in the CO record but will still be present in the SQ record of the clean coordinate file. | Boolean value Yes/No | No |
| [-ccfoutdir] (Parameter 2) |
outdir | This option specifies the location of CCF files (clean coordinate files) (output). A 'protein clean cordinate file' contains protein coordinate and other data for a single PDB file. The files, generated by using PDBPARSE, are in CCF format (EMBL-like) and contain 'cleaned-up' data that is self-consistent and error-corrected. Records for residue solvent accessibility and secondary structure are added to the file by using PDBPLUS. | Output directory | ./ |
| -logfile | outfile | This option specifies tame of the log file for the build. The log file may contain messages about inconsistencies or errors in the PDB files that were parsed. | Output file | pdbparse.log |
| Additional (Optional) qualifiers | ||||
| -[no]ccfnaming | boolean | This option specifies whether to use pdbid code to name the output files. If set, the PDB identifier code (from the PDB file) is used to name the file. Otherwise, the output files have the same names as the input files. | Boolean value Yes/No | Yes |
| -chnsiz | integer | Minimum number of amino acid residues in a chain for it to be parsed. | Any integer value | 5 |
| -maxmis | integer | Maximum number of permissible mismatches between the ATOM and SEQRES sequences. | Any integer value | 3 |
| -maxtrim | integer | Max. no. residues to trim when checking for missing C-terminal SEQRES sequences. | Any integer value | 10 |
| Advanced (Unprompted) qualifiers | ||||
| (none) | ||||
| Associated qualifiers | ||||
| "-pdbpath" associated dirlist qualifiers | ||||
| -extension1 -extension_pdbpath |
string | Default file extension | Any string | ent |
| "-ccfoutdir" associated outdir qualifiers | ||||
| -extension2 -extension_ccfoutdir |
string | Default file extension | Any string | ccf |
| "-logfile" associated outfile qualifiers | ||||
| -odirectory | string | Output directory | Any string | |
| General qualifiers | ||||
| -auto | boolean | Turn off prompts | Boolean value Yes/No | N |
| -stdout | boolean | Write first file to standard output | Boolean value Yes/No | N |
| -filter | boolean | Read first file from standard input, write first file to standard output | Boolean value Yes/No | N |
| -options | boolean | Prompt for standard and additional values | Boolean value Yes/No | N |
| -debug | boolean | Write debug output to program.dbg | Boolean value Yes/No | N |
| -verbose | boolean | Report some/full command line options | Boolean value Yes/No | Y |
| -help | boolean | Report command line options and exit. More information on associated and general qualifiers can be found with -help -verbose | Boolean value Yes/No | N |
| -warning | boolean | Report warnings | Boolean value Yes/No | Y |
| -error | boolean | Report errors | Boolean value Yes/No | Y |
| -fatal | boolean | Report fatal errors | Boolean value Yes/No | Y |
| -die | boolean | Report dying program messages | Boolean value Yes/No | Y |
| -version | boolean | Report version number and exit | Boolean value Yes/No | N |
% pdbparse Parses PDB files and writes protein CCF files. Pdb entry directories [./]: structure Mask non-amino acid groups in protein chains that do not contain a C-alpha atom. [N]: Mask amino acids in protein chains that do not contain a C-alpha atom. [N]: Y Mask amino acid residues in protein chains with a single atom only. [N]: Clean protein structure coordinates file output directory [./]: Domainatrix log output file [pdbparse.log]: Processing /homes/user/test/data/structure/1cs4.ent Processing /homes/user/test/data/structure/1ii7.ent Processing /homes/user/test/data/structure/2hhb.ent |
Go to the output files for this example
| FILE TYPE | FORMAT | DESCRIPTION | CREATED BY | SEE ALSO |
| PDB file | PDB format. | Protein coordinate data in PDB format. | N.A. | N.A. |
| Clean coordinate file (for protein) | CCF format (EMBL-like format for protein coordinate and derived data). | Coordinate and other data for a single PDB file. The data are 'cleaned-up': self-consistent and error-corrected. | PDBPARSE | Records for residue solvent accessibility and secondary structure are added to the file by using PDBPLUS. |
|
| Program name | Description |
|---|---|
| aaindexextract | Extract amino acid property data from AAINDEX |
| allversusall | Sequence similarity data from all-versus-all comparison |
| cathparse | Generates DCF file from raw CATH files |
| cutgextract | Extract codon usage tables from CUTG database |
| domainer | Generates domain CCF files from protein CCF files |
| domainnr | Removes redundant domains from a DCF file |
| domainseqs | Adds sequence records to a DCF file |
| domainsse | Add secondary structure records to a DCF file |
| hetparse | Converts heterogen group dictionary to EMBL-like format |
| jaspextract | Extract data from JASPAR |
| pdbplus | Add accessibility and secondary structure to a CCF file |
| pdbtosp | Convert swissprot:PDB codes file to EMBL-like format |
| printsextract | Extract data from PRINTS database for use by pscan |
| prosextract | Processes the PROSITE motif database for use by patmatmotifs |
| rebaseextract | Process the REBASE database for use by restriction enzyme applications |
| scopparse | Generate DCF file from raw SCOP files |
| seqnr | Removes redundancy from DHF files |
| sites | Generate residue-ligand CON files from CCF files |
| ssematch | Search a DCF file for secondary structure matches |
| tfextract | Process TRANSFAC transcription factor database for use by tfscan |
FILE_OPEN my.file
my.file could not be opened for reading or writing. The file is ignored.
File
FILE_READ my.file
my.file could not be read. The file is ignored.
File
NO_OUTPUT my.file
No clean coordinate file was generated for my.file. This will happen if there was a FILE_READ error on the raw PDB file, or a NOSEQRES, NOATOM or NOPROTEINS error when reading the file.
File
FILE_WRITE my.file
my.file could not be written. The file is ignored.
File.
BADINDEX 1 (A)
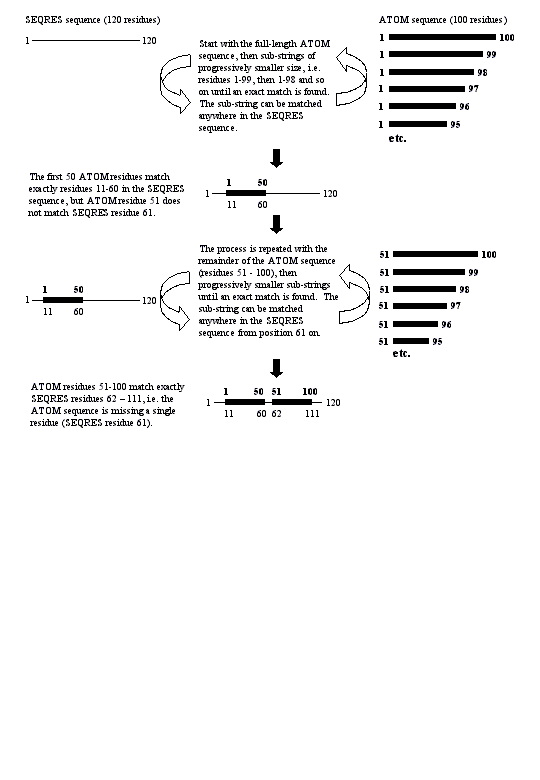
Raw residue numbering from ATOM records does not give the correct index into the SEQRES sequence for chain 1 ('A'). The correct alignment of the ATOM and SEQRES sequences is found by string handling (see 'Parsing methodology' in the text).
Chain
NEGNUM 1 (A) 123
Negative residue number found for chain 1 ('A') on line 123.
Chain
ZERNUM 1 (A) 123
Residue number of zero found for chain 1 ('A') on line 123.
Chain
ODDNUM 1 (A) 123
Possible residue heterogeneity or alternative residue numbering scheme for chain 1 ('A') on line 123.
Chain
NONSQNTL 1 (A) 123
Possible case of non-sequential numbering error for chain 1 ('A') on line 123.
Chain
HETEROK 1 (A)
Correct alignment of ATOM and SEQRES sequences of chain 1 ('A') found by presuming an alternative residue numbering scheme.
Chain
ALTERNOK 1 (A)
Correct alignment of ATOM and SEQRES sequences of chain 1 ('A') found by presuming one or more instances of heterogeneity.
Chain
MISSNTERM 1 (A) 3
SEQRES records appeared to be missing 3 N-terminal residues relative to ATOM sequence for chain 1 ('A'). The missing residues are added to the sequence.
Chain
MISSCTERM 1 (A) 3
SEQRES records appeared to be missing 3 C-terminal residues relative to ATOM sequence for chain 1 ('A'). The missing residues are added to the sequence.
Chain
GAPPEDOK 1 (A)
Correct alignment of ATOM and SEQRES sequences of chain 1 ('A') found by gap insertion with no mismatches.
Chain
MISMATCH 1 (A) 2 ALA 2 ARG 6; ALA 12 TYR 16
Correct alignment of ATOM and SEQRES sequences of chain 1 ('A') found without gap insertion but contained 2 mismatches (ALA 2 versus ARG 6 and ALA 12 versus TYR 16).
Chain
GAPPED 1 (A) 2 ALA 2 ARG 6; ALA 12 TYR 16
Correct alignment of ATOM and SEQRES sequences of chain 1 ('A') found by gap insertion but contained 2 mismatches (ALA 2 versus ARG 6 and ALA 12 versus TYR 16).
Chain
NOMATCH 1 (A)
Correct alignment of ATOM and SEQRES sequences of chain 1 ('A') could not be found by string handling (see 'Parsing methodology' in the text). The raw sequence from the ATOM records is taken to be the true sequence and the SEQRES sequence is discarded.
Chain
DUPATOMRES 3
Multiple sets of coordinates were given for an individual atom or whole residues, first instance on line 3. The first set of coordinates are used and the others discarded.
Residue
NOATOMRESID 123
No atom or residue identifier specified, first instance on line 123. All such lines are discarded.
Residue
SEQRESLENDIF 1(A)
Indicated and actual length of SEQRES sequence differs for chain 1 (chain identifier 'A'). The actual length of the sequence is used.
Chain
CHAINIDS 1 (A) 2 (A)
Chain identifiers of chains 1 and 2 are not unique ('A' in both cases). Both chains are discarded.
File
CHAINIDSPC
Space (' ') and non-space characters are both used for chain identifiers in a single file. Chains in ATOM records are identified by reference to the TER records as well as chain identifiers.
File
CHAINORDER 123
The order of the chains in the ATOM records is inconsistent with that in the SEQRES records, first instance on line 123. Coordinates are assigned to the correct chain by reference to the chain identifier.
File
TERNONE
No TER records were found. The chains in the ATOM records are identified by reference to the chain identifiers.
File
TERTOOMANY
Number of TER records is greater than the number of chains; possible digest.
File
TERTOOFEW
Number of TER records is less than the number of chains.
File
TERMISSHET 123 124
A chain is not separated from its heterogen group by a TER record between lines 123 and 124. Coordinates for the chain and heterogen are distinguished by reference to the chain identifier and residue numbers.
Chain
TERMISSCHN 123 124
Two chains are not separated by TER records between lines 123 and 124.
Chain
SEQRESNOAA 1 (A)
No known amino acids found in the SEQRES records for chain 1 ('A'). The chain is discarded.
Chain
SEQRESFEWAA 1 (A)
Fewer than the user-specified minimum number (5) of known amino acids were found in the SEQRES records for chain 1 ('A'). The chain is discarded.
Chain
NOPROTEINS
No chains were found with at least the user-specified minimum number (5) of known amino acids. The file is not parsed and no output file is generated.
File
ATOMFEWAA 1 (A) 3
Fewer than the user-specified minimum number of known amino acids found in the ATOM records for chain 1 ('A'), model 3. The chain is discarded.
Chain
SECMISS 123
One or more standard records (e.g. for residue identity) were missing for an SSE on line 123. The element(s) are discarded.
Line
SECBOTH 1 2 ALA 2 ARG 6
The start and end residues (ALA 2 ARG 6) of an element given in the HELIX, SHEET or TURN records was not found in the ATOM records of chain 1, model 2. The element is discarded.
Element
SECSTART 1 2 ALA 2
The start residue (ALA 2) of an element was not found in the ATOM records of chain 1, model 2. The element is discarded.
Element
SECEND 1 2 ARG 6
The end residue (ARG 6) of an element was not found in the ATOM records of chain 1, model 2. The element is discarded.
Element
SECCHAIN A
Chain identifier ('A') specified for an element not found in PDB file. The element is discarded.
Element
SECTWOCHN A B
2 chain identifiers ('A' and 'B') specified for an element. The element is discarded.
Element
NOSEQRES
No SEQRES records. The file is not parsed and no output file is generated.
File
NOATOM
No ATOM records. The file is not parsed and no output file is generated.
File
RESOLMOD
A value for the RESOLUTION record is given but MODEL records are also found. The file is presumed to contain an NMR structure or model.
File
NORESOLUTION
RESOLUTION record not found. The file is presumed to contain an NMR structure or model.
File
NOMODEL
NMR structure with no MODEL records. The number of models is determined by reference to the TER records.
File
MODELDUP 123
Duplicate MODEL records on line 123. The duplicate record is disregarded.
File
|
See also http://emboss.sourceforge.net/
For a publication that uses pdbparse,see for example Journal of Molecular and Cellular Cardiology 40 (2006) 234 - 236