This chapter will introduce you to a few of the EMBOSS applications that can be used to analyse protein sequences. Obviously, the pairwise sequence comparison methods illustrated in the previous chapter with nucleic acid sequences can also be used with protein sequences.
unix % plotorf
Plot potential open reading frames
Input sequence: embl:xlrhodop
Graph type [x11]:
You will see a graphical output that shows the potential open reading frames (ORF) in all six frames:
The longest ORF is in frame 2 from around position 100 to 1200. We will now identify the exact start and end points for our translation. To do this, we can use the EMBOSS program getorf.
unix % getorf -opt
Finds and extracts open reading frames (ORFs)
Input sequence: embl:xlrhodop
Output sequence [xlrhodop.orf]:
Genetic codes
0 : Standard
1 : Standard (with alternative initiation codons)
2 : Vertebrate Mitochondrial
3 : Yeast Mitochondrial
4 : Mold, Protozoan, Coelenterate Mitochondrial and Mycoplasma/Spiroplasma
5 : Invertebrate Mitochondrial
6 : Ciliate Macronuclear and Dasycladacean
9 : Echinoderm Mitochondrial
10 : Euplotid Nuclear
11 : Bacterial
12 : Alternative Yeast Nuclear
13 : Ascidian Mitochondrial
14 : Flatworm Mitochondrial
15 : Blepharisma Macronuclear
Code to use [0]:
Minimum nucleotide size of ORF to report [30]:
Type of sequence to output
0 : Translation of regions between STOP codons
1 : Translation of regions between START and STOP codons
2 : Nucleic sequences between STOP codons
3 : Nucleic sequences between START and STOP codons
4 : Nucleotides flanking START codons
5 : Nucleotides flanking initial STOP codons
6 : Nucleotides flanking ending STOP codons
Type of output [0]: 3
Notice that you can specify the organism whose codon usage table is most appropriate for your sequence, and you can also choose the type of information that is reported to you. In our case, we are simply interested in the positions of the start and stop codons for this sequence.
plotorf is just a graphical representation of the textual information produced by getorf. Since we asked for all ORFs above a minimum size to be reported, getorf is telling us about a number of potential ORFs. We know from plotorf that our ORF will be in the region 100 to 1200, so scroll through the output file, xlrhodop.orf, until you identify this. What are the actual start and end positions?
unix % more xlrhodop.orf
>XLRHODOP_7 [110 - 1171] Xenopus laevis rhodopsin mRNA, complete cds. atgaacggaacagaaggtccaaatttttatgtccccatgtccaacaaaactggggtggta cgaagcccattcgattaccctcagtattacttagcagagccatggcaatattcagcactg
From the previous exercise you should have found that the region to be translated is from 110 to 1171 in our cDNA sequence. Now we can use transeq to translate that region and use the translated peptide for some further analyses.
Let's practice using command line flags (qualifiers) again. The new ones here are
-sbegin and -send. These allow you to specify a
subregion of your sequence; in this case we will ask
transeq to translate only the part of
embl:xlrhodop that we have identified as the coding
region. You should remember -outseq from
Chapter 2.
unix % transeq embl:xlrhodop -sbegin 110 -send 1171 -outseq xlrhodop.pep
Translate nucleic acid sequences
unix % more xlrhodop.pep
>XLRHODOP+1 Xenopus laevis rhodopsin mRNA, complete cds. MNGTEGPNFYVPMSNKTGVVRSPFDYPQYYLAEPWQYSALAAYMFLLILLGLPINFMTLF VTIQHKKLRTPLNYILLNLVFANHFMVLCGFTVTMYTSMHGYFIFGQTGCYIEGFFATLG GEVALWSLVVLAVERYMVVCKPMANFRFGENHAIMGVAFTWIMALSCAAPPLFGWSRYIP EGMQCSCGVDYYTLKPEVNNESFVIYMFIVHFTIPLIVIFFCYGRLLCTVKEAAAQQQES ATTQKAEKEVTRMVVIMVVFFLICWVPYAYVAFYIFTHQGSNFGPVFMTVPAFFAKSSAI YNPVIYIVLNKQFRNCLITTLCCGKNPFGDEDGSSAATSKTEASSVSSSQVSPA
We saw earlier that the SwissProt entry for this protein has the identifier opsd_xenla; test your understanding of EMBOSS so far by using needle to compare your translated product with the database sequence. Compare your findings with the SwissProt entry using SRS.
| Residues 10-20 | sw:opsd_xenla[10:20] |
| The last ten residues | sw:opsd_xenla[-10:0] |
| The last twenty residues bar 5 | sw:opsd_xenla[-20:-6] |
| bases 134-458 reverse complement | embl:xlrhodop[134:458:r] |
The question of how DNA sequence determines specific protein structure has been a constant source of fascination and speculation since the problem was identified. It remains an extremely difficult area; generally referred to as the ``folding problem'', it is one of the major outstanding questions in molecular biology. Many attempts have been made to predict the tertiary structure of a protein from its sequence. These fall into two distinct approaches:
The approach to structure prediction based on mechanical models has the innate (possibly fatal) attraction that, in theory, it requires no prior knowledge of protein tertiary structure. If successful it could be applied uniformly to all sequences. By contrast, all methods based on inference from known structures are inherently limited in their applicability. They will only be appropriate for predicting structures similar to those which were used in the inference process. Fortunately there are often biophysical or biochemical clues that help make this decision and these are often integrated in the methods for structure prediction.
Currently the best way to achieve reasonable secondary structure predictions is to run a variety of prediction algorithms over your sequence and determine a consensus among the results. There are various web servers that will do these multiple analyses for you, including PIX at the HGMP and Jpred at the University of Dundee:
http://www.hgmp.mrc.ac.uk/Registered/Webapp/pix/
http://www.compbio.dundee.ac.uk/
 www-jpred
www-jpred
As yet, coverage of secondary structure prediction within EMBOSS is limited. More algorithms will be added to enable the conesensus approach described above. We'll take a look now at some of the predictions you can currently perform using EMBOSS.
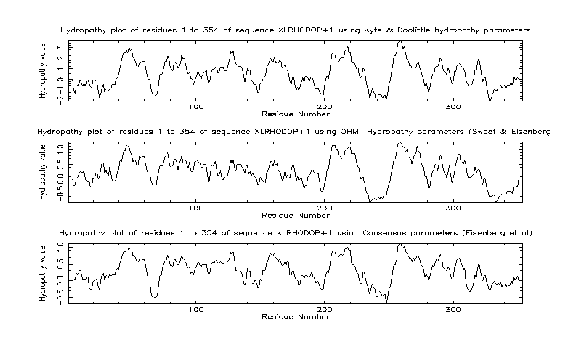
pepinfo produces information on amino acid properties (size, polarity, aromaticity, charge etc). Hydrophobicity profiles are also available and are useful for locating turns, potential antigenic peptides and transmembrane helices. Various algorithms are employed including the Kyte and Doolittle hydropathy measure - this curve is the average of a residue-specific hydrophobicity index over a window of nine residues. When the line is in the upper half of the frame, it indicates a hydrophobic region, and when it is in the lower half, a hydrophilic region.
unix % pepinfo xlrhodop.pep
Plots simple amino acid properties in parallel
Graph type [x11]:
Output file [pepinfo.out]:
You will see two screens (press return to move from the first
to the second screen) that look like this:
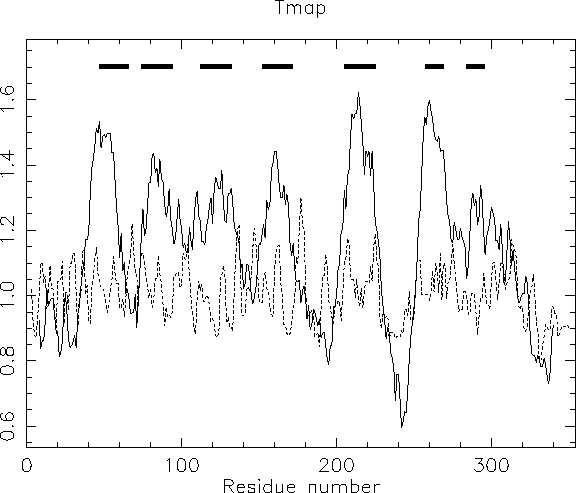
The results from the pepinfo hydropathy plot showed seven highly hydrophobic regions within xlrhodop.pep. Could these be transmembrane domains? We can use the EMBOSS program tmap to investigate this possibility:
unix % tmap
Displays membrane spanning regions
Sequences file to be read in: xlrhodop.pep
Graph type [x11]:
You will see a window that looks like this:
The bars across the top represent areas where transmembrane segments are predicted. Taken in combination with the results from pepinfo, we can see that there may be seven transmembrane helices in this protein. This corresponds well with both the SwissProt entry for this sequence (opsd_xenla) and with some information we will gather about patterns and profiles in the next chapter.
There are various other programs you can use to analyse your peptide sequence - to find out what is available, try rerunning wossname as we did in the first chapter.